Genome classifier
Implementation details
The classification of a fasta genome is divided into four steps:- Predict genes
We use Prodigal (Hyatt et al. BMC Bioinf 2010) to predict all genes and proteins from the input fasta file. - Extract Marker genes
We extract the 40 marker genes used to build the specI (Ciccareli et al. Science 2006, Mende et al. Nature Methods 2013) using fetchMG. - Map marker genes to the specI database
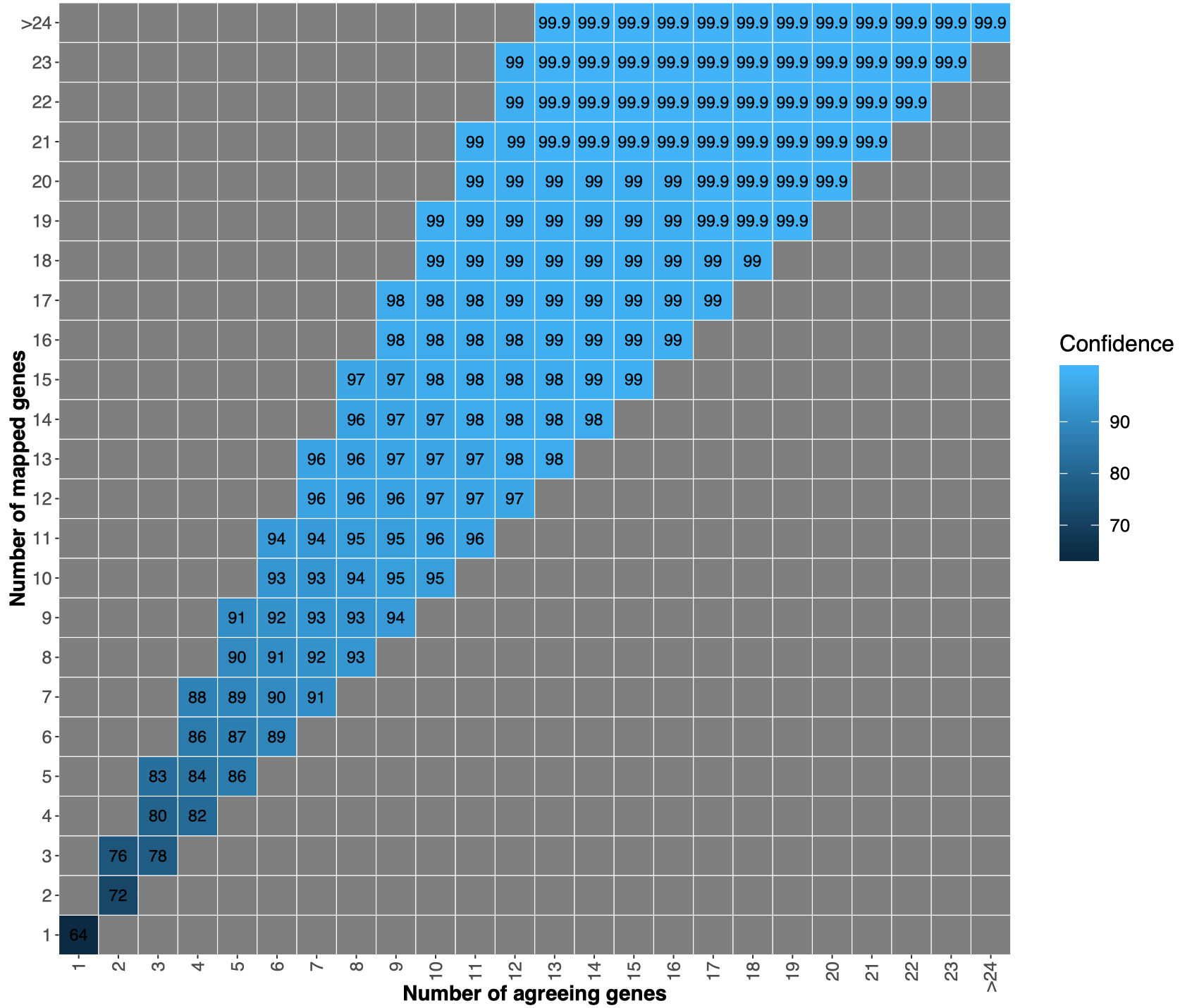
We map the extracted marker genes to the marker genes in the 12,226 specIs. We use vsearch (Rognes et al. PeerJ 2016) with the command--usearch_globaland option--maxrejects 10000 --minqt 0.7. We use marker gene specific thresholds that were calibrated in Milanese et al. Nature Comm. 2019 to distinguish between different species. For example,COG0049is a conserved gene with threshold98.9, whileCOG0124is evolutionary under less pressure having a threshold of94.7. - Find taxonomy annotation for the genome
From the annotation of the single genes we derive the annotation for the submitted genome, which can be:No genes, no marker genes have been detected in the fasta file;No match, marker genes have been detected in the input genome, but there is no match to the specI taxonomy;Inconsistent, marker genes have been detected and map to the specI database, but half of the genes map to one specI and the other half to another specI;Consistent, the majority of the genes agree on one specI.
Consistent matches, we did a 10-fold cross-validation.